longrnaseq
Introduction
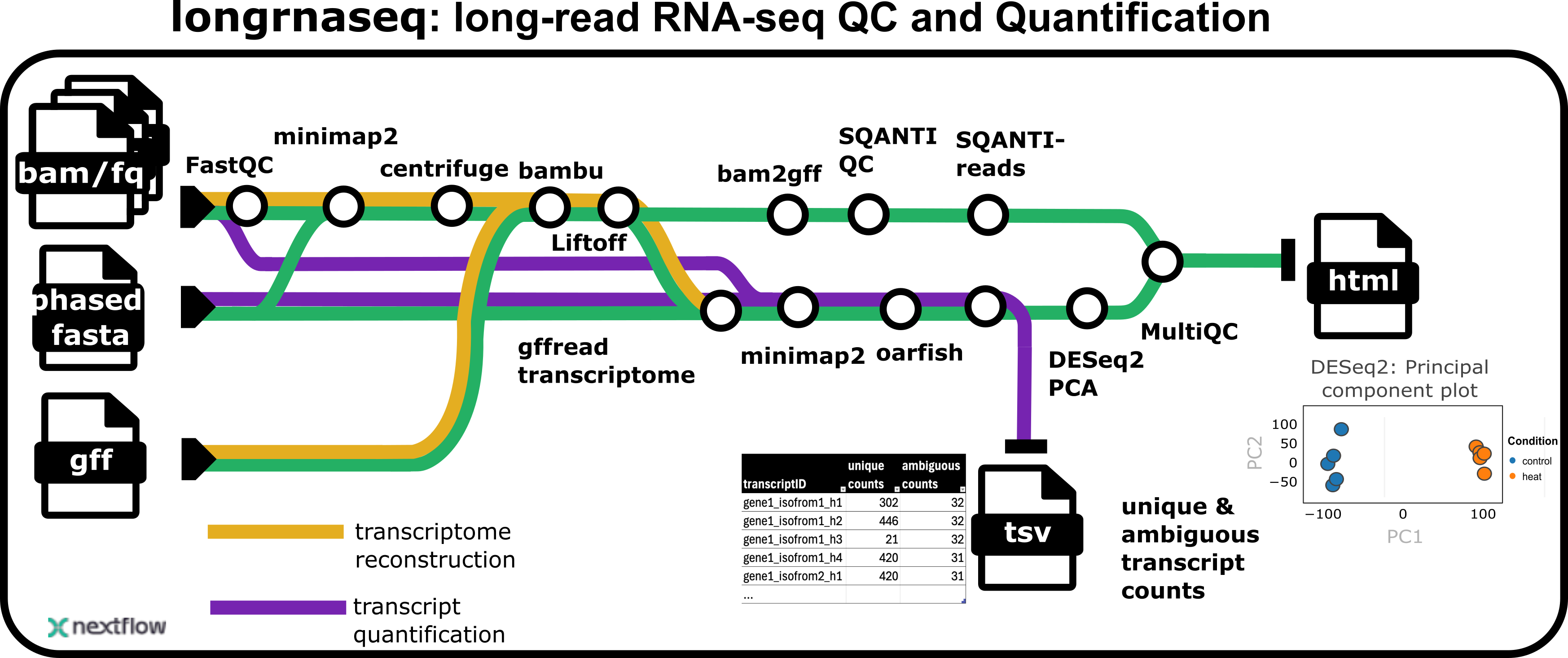
longrnaseq is a bioinformatics pipeline that processes long-read RNA sequencing data. The pipeline performs quality control, alignment, classification, contamination detection, and transcript quantification for long-read RNA-seq data from multiple samples.
The pipeline includes the following main steps:
Read QC (FastQC)
Present QC for samples (MultiQC)
Genome alignment (Minimap2)
Contamination detection (Centrifuge)
Comparion of samples by transcript classification (SQANTI-reads)
Transcript quantification (Oarfish) and gene-level summarization
Dependencies
An environment with nextflow (>=24.04.2) and Singularity installed.
Note: If you want to run SQANTI-reads quality control, you will also need to:
Install all SQANTI3 dependencies in the same environment as nextflow/nf-core environment (sorry there is not functional container for nextflow at the moment..)
Important: for converting output to html poppler also need to be installed: conda install poppler
Clone the SQANTI3 git repository and provide the directory as input. v ==5.5.4
For running Centrifuge, you also need to create a Centrifuge database.
Both of these can be skipped with --skip_sqanti and --skip_centrifuge
Usage
Clone the repository of the pipeline
git clone https://github.com/nadjano/longrnaseq.gitPrepare a samplesheet with your input data that looks as follows:
samplesheet.csv:
sample,fastq_1
SAMPLE1,sample1.fastq.gz
SAMPLE2,sample2.fastq.gz
Each row represents a sample with one fastq file.
Running the Pipeline
Required Parameters
The pipeline requires the following mandatory parameters:
--input: Path to samplesheet CSV file--outdir: Output directory path--fasta: Path to reference genome FASTA file--gtf: Path to GTF annotation file (for BAMBU to get the right output with gene_id!)--centrifuge_db: Path to Centrifuge database--sqanti_dir: Path to SQANTI3 directory--technology: ONT or PacBio, sets minimap2 parameters for read mapping
Note about gtf file
gtf-version 3
should include features: gene, transcript, exon, CDS
Profile Support
Currently, only the singularity profile is supported. Use -profile singularity in your command.
Example Command
nextflow run main.nf -resume -profile singularity \
--input assets/samplesheet.csv \
--outdir results \
--fasta /path/to/genome.fa \
--gtf /path/to/annotation.gtf \
--centrifuge_db /path/to/centrifuge_db \
--sqanti_dir /path/to/sqanti3 \
--technology ONT/PacBio \
Optional Parameters
--skip_deseq2_qc: Skip deseq2, when only one sample is present deseq2 will fail [default: false]--skip_sqanti: Skip sqanit and sqanti reads [default: false]--skip_centrifuge: Skip centrigure [default: false]-bg: Run pipeline in background-resume: Resume previous run from where it left off--downsample_rate: fraction between 0-1 for downsampling before running SQANTI3 to reduce runtime and for vizualization to have smaller files [default: 0.05]--large_genome: In case minimap2 fails druing genome indexing, this can be due to large genomes and long chromosomes. [default: false]
Pipeline output
The main output is a MultiQC.html and oarfish transcript and gene counts.
An example MultiQC report can be found here
Running on HPC
For running the pipeline on a HPC (e.g SLURM) you need to add some configuartion to the nextflow.config file
e.g:
process.executor = 'slurm'
process.clusterOptions = '--qos=short' # if you have to submit to a specific queue
Test Run
A test dataset is available for testing and demonstration purposes. This dataset contains a phased genome assembly and annotation for chromosome 1 across all haplotypes of the tetraploid potato cultivar Atlantic.
long-read RNA-seq fastq files:
Download from SRA the samples: SRR14993893 and SRR14993894.
First add samples to sample sheet, download the annotation files and then run the pipeline like this:
nextflow run main.nf -profile singularity \
--input assets/samplesheet.csv \
--outdir output_test \
--fasta test_data/ATL_v3.asm.with_chloroplast_and_mito.fa \
--gtf test_data/unitato2Atl.with_chloroplast_and_mito.no_scaffold.agat.gtf \
--technology ONT --downsample_rate 0.99 --skip_centrifuge --skip_sqanti -resume
This should finish in less than one hour (running with 30 cpu) including pulling of singularity images.
Contributions and Support
If you would like to contribute to this pipeline, please get in touch nadja.franziska.nolte[at]nib.si
Citations
An extensive list of references for the tools used by the pipeline can be found in the CITATIONS.md file.
You can cite the nf-core publication as follows:
The nf-core framework for community-curated bioinformatics pipelines.
Philip Ewels, Alexander Peltzer, Sven Fillinger, Harshil Patel, Johannes Alneberg, Andreas Wilm, Maxime Ulysse Garcia, Paolo Di Tommaso & Sven Nahnsen.
Nat Biotechnol. 2020 Feb 13. doi: 10.1038/s41587-020-0439-x.