Syntelog identification in hexaploid wheat
This is an example of running the syntelogfinder pipeline on an example. We will use a wheat long-read RNA-seq dataset from the cultivar AK58.
Part 1: Preparing the Phased Reference Genome
gff: https://download.cncb.ac.cn/gwh/Plants/Triticum_aestivum_1_GWHANRF00000000/GWHANRF00000000.gff.gz
The chromosome names are not so nice, so we will rename them:
e.g. GWHANRF00000001 –> chr1_A
e.g. GWHANRF00000002 –> chr1_B
e.g. GWHANRF00000003 –> chr1_C
Now we are ready to run the syntelog finder pipeline.
Part 2: Running the Syntelog Finder Pipeline
Install nextflow and conda
Prepare the params.config file
params/wheatAK58.json
{
"reference_fasta": "/scratch/nadjafn/LR_DESIREE_PAPER/ANALYSIS/wheat_example/genome/GWHANRF00000000.renamed.fasta",
"reference_gff": "/scratch/nadjafn/LR_DESIREE_PAPER/ANALYSIS/wheat_example/genome/GWHANRF00000000.renamed.gff",
"ploidy": 3,
"outdir": "/DKED/scratch/nadjafn/potato-allelic-orthogroups/output_wheat"
}
nextflow run main.nf -resume -params-file params/wheatAK58.json -c conf/nextflow.config -profile conda -bg
Why ploidy 3? It is a hexaploid species but we only have A, B and D subgenomes.
Results
The main output we are interested in is the syntelogfinder/output_wheat/03_GENESPACE directory, which contains these three files:
GWHANRF00000000.renamed_genespace.pie_chart.svg

GWHANRF00000000.renamed_genespace_combined_barplots.svg

We can see here that the exon lengths are very different between the genes in the 1hapA_1hapB_1hapD_s synteny category, but the exon lengths are more similar within each haplotype, with most of them having the same lengths.
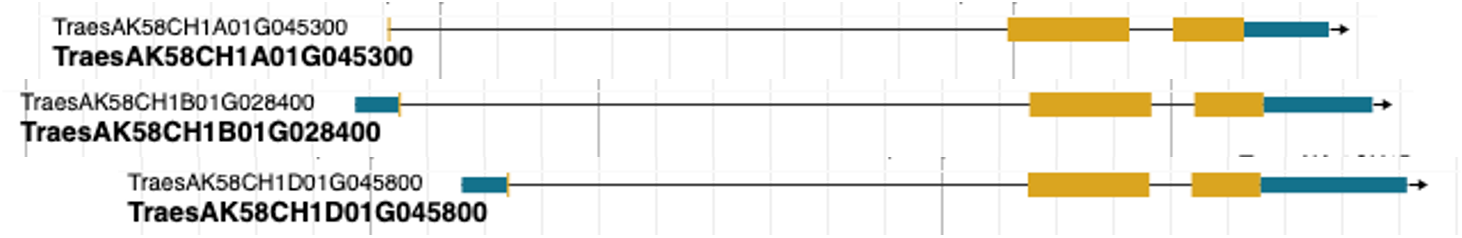
So to avoid any bias in read mapping to the longest haplotype (if on the other haplotypes the transcript is too short) we will modify the gff3 file to “chop” the UTRs off that more transcripts have the same length.
Part 3: Long-Read RNA-Seq Analysis
Prepare the assets/sample.csv file:
.. code-block:: text
sample,fastq_1 SRR33004955,fastq/SRR33004955.fastq SRR33004956,fastq/SRR33004956.fastq SRR33004957,fastq/SRR33004957.fastq SRR33004958,fastq/SRR33004958.fastq
The wheat is very large so we need to use the option --large_genome to choose the right mapping options.
nextflow run main.nf -resume -profile singularity \
--input assets/samplesheet_AK58.csv \
--outdir output_wheat_AK58 \
--fasta genome/GWHANRF00000000.renamed.fasta \
--gtf GWHANRF00000000.renamed.cds2exon.gtf \
--centrifuge_db centrifuge/dbs_v2018/ \
--sqanti_dir sqanti3/release_sqanti3 \
--sqanti_test -bg --technology PacBio --large_genome